| |
KEYNOTE AND PLENARY LECTURE ABSTRACTS
Playing Practical Games with Bacteria and Viruses. Exploring the Molecular Mechanisms behind Clever Cellular Stratagems
Adam Arkin
University of California, Berkeley, CA
How do pathogenic bacteria sense their environment to deploy different survival strategies? Why do some viruses, like HIV, allow their host to live for long periods whereas others like Ebola do not? How precisely are these strategies encoded in the organism's biochemistry and genetics and how closely do they need to be followed to guarantee its survival? What are the optimal strategies for defeating these organisms or forcing them to do our bidding for industrial or medical benefit? Here I will demonstrate, using examples from our research on Bacillus subtilis stress response and the design of HIV gene therapeutic strategies, how molecular biology combined with methods from statistical physics, nonlinear dynamics, and game theory can be used to pose and partially answer these questions as well as illustrate some of the profound challenges in doing so.
A Genome-scale Protein Interaction Map for Drosophila melanogaster
Joel Bader
John Hopkins University, Baltimore, MD
Drosophila melanogaster is a proven model system for many aspects of human biology. Here we present a two-hybrid-based protein-interaction map of the majority of the fly proteome. 10,623 predicted transcripts were isolated and screened against standard and normalized cDNA libraries to produce a draft map of 7048 proteins and 20,405 interactions. A computational method of rating two-hybrid interaction confidence was developed to refine this draft map to a higher confidence map of 4679 proteins and 4780 interactions. Statistical modeling of the network showed two levels of organization: a short range organization, presumably corresponding to multiprotein complexes and a more global organization, presumably corresponding to inter-complex connections. The network recapitulated known pathways, extended pathways, and uncovered novel pathway components. This map serves as a starting point for a systems biology modeling of multicellular organisms including humans.
The architecture of complexity: Structure and modularity in cellular networks
Albert-Laszlo Barabasi
University of Notre Dame, Notre Dame, IN
Post genomic biology requires us to move beyond the single gene
description, and understand the intricate genetic networks that mediate
most cellular processes.
In the last few years we learned that cellular networks are not
random, but their structure carries the signature of self-organizing
processes governed by simple but generic laws.
The analysis of the metabolic network of 43 organisms and of the
protein interaction network of the yeast indicate that, despite
significant variances in their individual components, these networks
display identical topologic and scaling
properties, being described by scale-free networks. The hubs, highly
connected nodes common in such networks, have important
implications on the cell's robustness and functionality. Finally, I will
show that cellular networks have a hierarchical architecture, allowing us to
identify the organization of the functional modules embedded in the
cellular topology.
For more information see http://www.nd.edu/~networks
Integrative modeling of mRNA expression and transcription factor binding data
Harmen Bussemaker
Columbia University, New York, NY
Functional genomics studies are yielding information about regulatory processes in the biological cell at an unprecedented scale. Not only have DNA microarrays been used to measure, for all genes simultaneously, the mRNA abundance in a variety of conditions, but the level of occupancy of their promoter regions by a large number of transcription factors has also been determined. The challenge is to extract useful information about the global regulatory network from these data. We present an integrative modeling framework that combines libraries of expression and occupancy data to define the functional targets of each transcription factor: Multivariate regression analysis is used to infer transcription factor activity levels for each condition, and the correlation between the mRNA expression profile of an individual gene and the inferred activity profile of a transcription factor is interpreted as regulatory coupling strength. Applying our method for the yeast S. cerevisiae, we find that on average 58% of the genes whose promoter region is bound by a transcription factor are true regulatory targets. Moreover, our results enable us to assign directionality to transcription factors controlling divergently transcribed genes that share the same promoter region. These findings have implications for the global modeling of regulatory networks.
Network Analysis of Gene Expression Time Series
Greg Dewey
Keck Graduate Institute, Claremont, CA
There has been considerable interest in computational techniques for inferring genetic regulatory networks from whole-genome expression profiles. When expression time series data sets are available, dynamic models can, in principle, be used to determine correlative relationships between gene expression levels. Network models derived from simple rate laws offer an intermediate level analysis, going beyond simple statistical analysis, but falling short of a fully quantitative description. This talk discusses how such network models can be constructed and describes the global properties of the networks derived from such a model. These global properties are statistically robust and provide insights into the design of the underlying network. Several whole-genome expression time series datasets from yeast microarray experiments were analyzed using a Markov-modeling method to infer an approximation to the underlying genetic network. We found that the global statistical properties of all the resulting networks are similar. The overall structure of these biological networks is distinctly different from that of other recently studied networks such as the Internet or social networks. In addition to the small world properties, the biological networks show a power law or scale free distribution of connectivities. An inverse power law, 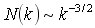 , for the number of vertices (genes) with k connections was observed for three different data sets from yeast. We propose network growth models based on gene duplication events that closely mimic the experimental derived networks. , for the number of vertices (genes) with k connections was observed for three different data sets from yeast. We propose network growth models based on gene duplication events that closely mimic the experimental derived networks.
Riboswitches: possibly the oldest regulatory system
Mikhail Gelfand
State Scientific Center GosNIIGenetika, Moscow, Russia
Riboswitches are RNA structures in mRNAs that directly bind small ligands and regulate gene expression in a number of metabolic systems. The remarkable features of riboswitches are conservation on very large phylogenetic distances (all major branches of eubacteria, archaea, eukaryotes) and their involvement in different regulatory interactions (attenuation of transcription, attenuation of translation, inhibition of translation initiation) leading to either activation or repression of gene expression. This combination of independence of other factors, functional diversity and sequence conservation argues for very ancient origin of these structures. Known riboswitches regulated metabolism of vitamins (riboflavin, cobalamin, thiamin), amino acids (methionine and lysine) and purines.
Computational analysis has played a major role in the discovery of riboswitches, dissecting their mechanism of action, and functional annotation of genes regulated by riboswitches. In combination with other comparative genomic techniques such as analysis of positional clusters and phylogenetic patterns, analysis of riboswitches and other regulatory elements allowed us to identify new transporters of riboflavin, thiamin and its metabolic precursors, cobalamin, nickel, lysine, methionine, as well as a number of enzymes involved in the corresponding metabolic pathways. Several such predictions have already been confirmed in experiment. We also studied the evolution of regulatory networks that involve riboswitches, and some examples of such networks will be presented in the talk.
Scale-free, small-world interaction networks: applications for function prediction and a neutral scenario for their evolution.
Martijn A. Huynen
University of Nijmegen, Nijmeegse, Netherlands
We study the properties of networks of predicted interactions between
proteins. Networks based on the conservation of gene-order in prokaryotes
as well as on co-expression in eukaryotes display a small-world,
scale-free architecture with a high degree of local clustering. In terms
of the application for function prediction the high degree of local
clustering implies that we obtain a "comparative-genomics" based
definition of functional modules: sets of proteins that are involved in a
single task. Co-expression networks are shown to be similar to gene-order
networks in the sense that conservation of co-expression increases the
likelihood of functional interaction between the proteins. In terms of
their evolution we study the minimal conditions under which a small-world,
scale-free architecture with a high degree of local clustering can evolve.
We show that a simple mechanistic model can account for the architecture
of the network. In this model 1) genes are duplicated with their
transcription factor binding sites (TFBS), 2) individual TFBS can be
duplicated and lost, 3) genes can be lost and 4) genes that share multiple
TFBS are co-expressed and connected in the network. This model is
supported by independent data on the sharing of transcription factor
binding sites between paralogs and on the levels of co-expression between
paralogs. The simplicity and neutrality of the model question arguments
about the benefit of the small-world, scale-free architecture for
intracellular organisation.
Graph Theory and Community Structure in Infectious Disease Models
M. Elizabeth Halloran
Emory University, Atlanta, GA
Transmission of infectious diseases
within communities depends on the contact patterns of individuals within
the community. Transmission units such as households, schools,
workplaces, and hospitals play an important role. In this talk, we
discuss our stochastic simulation model of smallpox transmission and the
construction of the community structure. We apply graph theoretic
considerations to compare the community structure in our simulator with
the community structure in other simulators. We use the degree
distribution, clustering coefficient distribution, shortest path
distribution, among others to make the comparison. The different
simulators produce qualitatively similar results of interventions
against smallpox. The insights gained from graph theory has potential
for developing general mixing structures in infectious disease
transmission models. This is joint work with Ira Longini and Azhar Nizam
of Emory University, and Stephen Eubank, Madhav Marathe, Rahul Tripathi,
Anil Kumar, and Nicholas Hengartner of Los Alamos National Laboratory.
A Comprehensive Set of Protein Complexes by Cluster Analysis of High Throughput Biochemical Purifications in Yeast
Roland Krause
Cellzome, Heidelberg, Germany
The analysis of protein?protein interactions allows for detailed
exploration of the cellular machinery and has become one of the most
important tools for the elucidation of protein function. The biochemical
purification of protein complexes followed by identification of components
by mass spectrometry is currently the method, which delivers the most
reliable information ? albeit that the data sets are still difficult to
interpret. Consolidating individual experiments into protein complexes for
high-throughput screens has been useful to reduce the complexity and to
facilitate annotation and interpretation of results.
Because of contaminants, the occurrence of proteins in otherwise
dissimilar purifications due to functional re-use, and technical
limitations in the detection these procedures were only carried out mainly
manually so far, even for high throughput screens. It would be beneficial
to have an procedure at hand to consolidate the experiments automatically.
I will present a measure to define similarity within collections of
purifications and generate a set of minimally redundant, comprehensive
complexes using unsupervised clustering. The method can be used on
different data sets and produces biologically meaningful complexes.
Simple stochastic birth and death models of genome evolution: Was there enough time for us to evolve?
Eugene V. Koonin
National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, MD
The distributions of many genome-associated quantities, including the membership of paralogous gene families can be approximated with power laws. We are interested in developing mathematical models of genome evolution that adequately account for the shape of these distributions and describe the evolutionary dynamics of their formation.
We show that simple stochastic models of genome evolution lead to power law asymptotics of protein domain family size distribution. These models, called Birth, Death and Innovation Models (BDIM), represent a special class of balanced birth-and-death processes, in which domain duplication and deletion rates are asymptotically equal up to the second order. The simplest, linear BDIM shows an excellent fit to the observed distributions of domain family size in diverse prokaryotic and eukaryotic genomes. However, the stochastic version of the linear BDIM explored here predicts that the actual size of large paralogous families is reached on an unrealistically long timescale. We show that introduction of non-linearity, which might be interpreted as interaction of a particular order between individual family members, allows the model to achieve genome evolution rates that are much better compatible with the current estimates of the rates of individual duplication/loss events.
BPPS: An algorithm for analyzing protein sequence alignments
Jun Liu
Harvard University, Boston, MA
Aligning multiple biopolymer sequences has been recognized as a central
activity in bioinformatics research. But the analysis of the resulting
alignments has not been rigorously formulated and mathematically tackled.
We have developed statistical procedures to decompose the multiple
alignment into distinct categories and to pinpoint critical structural
features within each category. A central part of our statistical
procedures is a novel algorithm called the Bayesian partitioning with
pattern selection (BPPS), which is based on a two-way mixture model and
can simultaneously classify protein sequences into distinct subfamilies
and select conserved positions that are characteristic of these
subfamilies. When applied to P-loop GTPases, this revealed within
Rab, Rho, Ras, and Ran a canonical network of molecular interactions
centered on bound nucleotide. This network presumably performs a crucial
structural and/or mechanistic role considering that it has persisted for
more than a billion years after the divergence of these families.
Detecting topological patterns in protein networks
Sergei Maslov
Brookhaven National Laboratory, Upton, New York
Bio-molecular networks lack the top-down design.
Instead, selective forces of biological evolution
shape them from raw material provided by random events
such as gene duplications and single gene mutations.
As a result individual connections in these networks
are characterized by a large degree of randomness.
One may wonder which connectivity patterns are indeed random,
while which arose due to the network growth, evolution, ans/or its
fundamental design principles and limitations?
Here we introduce a general method [1,2,3] allowing one to
construct a random version of a given network
while preserving the desired set of its low-level
topological features, such as, e.g., the number
of neighbors of individual nodes, the average
level of modularity, preferential connections
between particular groups of nodes, etc.
Such a null-model network can then be used to detect
and quantify non-random topological patterns.
In particular, we measure correlations between connectivities of
interacting nodes in protein interaction and regulatory networks in
yeast [1]. It was found that in both these networks,
links between highly connected proteins are
systematically suppressed. This effect decreases the likelihood of
cross talk between different functional modules of the cell,
and increases the overall robustness of a network by localizing
effects of deleterious perturbations.
We proceed by presenting the set of empirical results on
how gene duplications shape protein interaction
and genetic regulatory networks [4]. It is shown that
evolving molecular networks combine the upstream
plasticity of transcriptional regulation with the
downstream robustness of the protein function.
We believe this to be a general feature affecting
the evolvability of bio-molecular networks.
1. S. Maslov and K. Sneppen, Specificity and Stability in Topology of Protein Networks, Science 296, 910-913, (2002).
2. S. Maslov, K. Sneppen, and A. Zaliznyak, Pattern Detection in Complex Networks: Correlation Profile of the Internet, Preprint at arXiv.org e-Print archive available at http://arxiv.org/abs/cond-mat/0205379, (2002); Physica A, in press (2003).
3. The set of MATLAB programs can be downloaded from
http://www.cmth.bnl.gov/~maslov/matlab.htm
4. S. Maslov, K. Sneppen, and K. Eriksen, Upstream Plasticity and Downstream Robustness
in Evolution of Molecular Networks. Quantitative biology e-Print archive http://arxiv.org/archive/q-bio (2003).
Protein Complexes and Functional Modules in Molecular Networks
Leonid Mirny
Massachusetts Institute of Technology, Cambridge, MA
Proteins, nucleic acids, and small molecules form a dense network of molecular interactions in a cell. Molecules are nodes of this network and the interactions between them are edges. The architecture of molecular networks can reveal important principles of cellular organization and function, similarly to the way that protein structure tells us about the function and organization of a protein. Computational analysis of molecular networks has been primarily concerned with node degree or degree correlation, and hence focused on single/two-body properties of these networks. Here, by analyzing the multi-body structure of the network, we discovered molecular modules that are densely connected within themselves but sparsely connected with the rest of the network. Comparison with experimental data and functional annotation of genes showed that such modules correspond either to protein complexes (splicing machinery, transcription factors, etc.) or to dynamic functional units (signaling cascades, cell-cycle regulation, etc.). These modules are highly statistically significant, as is evident from comparison with random graphs, and are robust to noise in the data. Our results provide strong support for the network modularity principle introduced by Hartwell et al, suggesting that the found modules constitute the "building blocks" of molecular networks.
Conserved networks of interactions within protein structures
Shmuel Pietrokovski
Weizmann Institute of Science, Israel
Proteins are complex biological molecules that have highly diverse functions. The structure adopted by each protein chain is crucial for its function. We represent protein structures as networks of amino acids. Edges connect amino acids that physically interact with each other. This interaction can be characterized its type (hydrogen bond, hydrophobic interaction etc.) and weighted by different parameters (distance, contact surface area, energy etc.). This novel description of proteins offers new ways for their analysis. By analyzing thousands of distinct structures we found characterizing features of each node type (amino acid). We can use these features to identify atypical nodes in protein structure networks. Comparing the networks of homologous proteins identifies the conserved network regions. These indicate the interactions and amino acids important for the protein function, such as its active site. Finally, we found that active sites can also be identified by network characters of the nodes.
On Truth, Pathways and Interactions
Andrey Rzhetsky
Columbia University, New York, NY
I will give an overview of our effort to automatically extract pathway information from a large number of full-text research articles (GeneWays system), automatically curate the extracted information, and to combine the literature-derived information with sequence and experimental (such as yeast two-hybrid) data using a probabilistic approach.
Function, Design, and Construction of Gene Circuitry
Michael A. Savageau
University of California, Davis, CA
The ability to comprehensively and quantitatively monitor dynamic
changes in gene expression, together with new genome-scale informatic
methods, is enabling high-throughput characterization of genetic
regulatory networks. In addition, methods of genetic engineering now
allow synthetic regulatory circuits to be readily built. Attention
is currently being turned towards manipulating genetic regulatory
circuits for therapeutic and technological applications, which
increases the need to understand the functional consequences of
genetic manipulations and to discover principles that can guide the
design process. This issue will be addressed by comparing and
contrasting what has been learned about natural gene circuits in
their complex natural setting and what has been learned from
designing, constructing and analyzing simple synthetic gene circuits.
Imprint of Evolution on Protein Structural Universe
Eugene Shakhnovich
Harvard University, Cambridge, MA
The analysis of uneven fold populations in the universe of protein domains with the use of graph theory lead to the discovery of striking relation between analogous folds in the universe of protein domains whereby it is organized into a scale-free network in number of structurally-related folds, in contrast with a control random graph. We show, using phenomenological gene duplication models that such scale-free character of protein domain universe may be an imprint of Big Bang scenario of protein genesis where most of the folds emerged from one or few precursors via divergent evolution that includes gene duplications and mutations. Further, in order to get a more microscopic insight into the evolutionary origin of diverse protein folds and scale-free nature of protein structural universe we directly simulated divergent evolution of protein domains within three-dimensional lattice model. While missing many details of real proteins, this model nevertheless captures specific sequence-structure relationship - a key ingredient for modeling of protein structural evolution. Starting from an initial seed structure, the evolution of model proteins progresses by gene duplication with subsequent point mutations. A new gene's ability to fold and be stable in a unique structure is tested each time in direct kinetic folding simulations. In case it can, the algorithm accepts the new sequence and structure and thus a new protein is born. In each run the model evolution provides several thousand new model proteins with diverse structures. Analysis of evolved structures shows that they are much more designable than original structures as judged by recently developed (England and Shakhnovich, 2003) structural determinant of protein designability and direct sampling of their sequence spaces. We test the prediction drawn from model evolution on real proteins and show that protein domains that are found in eukaryotic organisms only and thus perceived as later evolved ones feature statistically significant higher designability than their prokaryotic counterparts. These results present a fundamental view on protein evolution highlighting relative role of structural selection and evolutionary dynamics in genesis of modern proteins.
Emergence and selection in Genome Evolution
Richard Solé
Universitat Pompeu Fabra, Barcelona, Spain
Recent models of genome-proteome evolution have shown that some of the
key traits displayed by the global structure of cellular networks might
be a natural result of a duplication-diversification process. One of the
consequences of such evolution is the emergence of a small world architecture
together with a scale-free distribution of interactions and modular structure.
Although these models lack any functionality and are thus free from
meeting functional constraints, they display the observed features observed in
real proteome maps, when tuned close to a phase transition point separating a
highly connected graph from a disconnected system. Close to such boundary,
the maps are shown to be scale-free hierarchical organization, behave as small
worlds and exhibit modularity.
It is conjectured that natural selection tuned the average connectivity in such
a way that the network is optimal at low cost (sparse graph). One consequence of such scenario is that the scaling laws and the essential ingredients for
building a modular net emerge "for free" close to such transition.
Orthologous protein domains in eukaryotes
Erik.Sonnhammer
Karolinska Institutet, Stockholm, Sweden
One of the most reliable methods for protein function
annotation is to transfer experimentally known functions from
orthologous proteins in other organisms. Most methods for identifying
orthologs operate on a subset of organisms with a completely sequenced
genome, and treat proteins as single-domain units. However, it is
well known that proteins are often made up of several independent
domains, and there is a wealth of protein sequences from genomes that
are not completely sequenced. We have developed a database of
orthologous protein domains based on Pfam domain families called HOPS:
Hierarchy of Orthologous and Paralogous Sequences. Orthology is
inferred by the Orthostrapper algorithm in a hierarchical system of
distinct phylogenetic subgroups. The results are accessible in the
graphical browser NIFAS that displays gene trees, domain
architectures, and orthology relationships. The method was tested on a
set of curated orthologs with experimentally verified function. In
comparison to tree reconciliation with a complete species tree, our
approach finds significantly more orthologs in the testset. Examples
for investigating gene fusions and domain recombination using HOPS
will be presented.
Computational Cell Biology: From Molecular Networks to Cell Physiology
John J. Tyson
Virginia Polytechnic Institute and State University
The fundamental goal of molecular cell biology is to understand cell
physiology in terms of the information encoded in the cell's genome. In
principle, we know how this information is translated into functional
proteins that carry out most of the interesting chores in a living cell.
But to make a firm connection between molecular events and cell behavior
involves many challenging computational problems at every step along the
way. The early steps--sequence analysis, protein folding, molecular
dynamics, metabolic control theory--are well established branches of
biochemistry. But the 'last step', from networks of regulatory proteins to
the physiological responses of a cell to its environment, is an especially
challenging problem that has received little attention so far. Accurate and
effective computational methods for deriving cell behavior from molecular
wiring diagrams are crucial to future progress in understanding living
cells and in modifying cell physiology for medical and technological purposes.
A nice example of this challenge is the cell cycle: the sequence of events
by which a growing cell duplicates all its components and partitions them
more-or-less evenly between two daughter cells. The cell cycle is
fundamental to all processes of biological growth, development and
reproduction, and hence plays a central role in such important processes as
carcinogenesis, wound healing, and tissue engineering. The molecular
mechanism that controls DNA synthesis and nuclear division is so complex
that its behavior cannot be understood by casual, hand waving arguments. By
translating this mechanism into differential equations, we can analyze and
simulate the behavior of the control system, comparing model predictions to
the observed properties of cells. Theoretical models also provide new ways
to look at the dynamics of cell cycle regulation. This approach is
generally applicable to any complex gene-protein network that regulates
some behavior of a living cell.
The Optimality, Expression, and Robustness of Cellular Metabolic Networks
Dennis Vitkup
Harvard Medical School, Boston, MA
Availability of whole-cellular networks allows one to study the global properties of system optimality, expression, and robustness. I will present our recent results which shed light on these important properties for the metabolic networks of E.coli and S. cerevisiae. Using an analysis of the biochemical flux distribution in the networks, we investigate the optimality of the natural and perturbed metabolic networks. We show that two general principles governing the network behavior are the optimality in the native state and prevalence of the homeostatic regulation upon perturbations. We demonstrate how these properties could be used to predict the re-distribution of fluxes. Using in-silico gene deletions and the optimization of flux distribution we estimate the contribution of the network structure to the system robustness against deleterious mutations. Towards the goal of understanding dynamic properties of cellular metabolic networks we investigate the regulation of gene expression in S. cerevisiae. We find that the gene correlation in the network is local in nature. The co-expression generally extends to distances equal to the average gene network connectivity. A significant fraction of the S. cerevisiae biochemical pathways display statistically significant gene co-expression and gene co-expression in linear pathways is significantly higher compared to non-linear pathways. We investigate the characteristic co-expression patterns exhibited by local topological motifs in the network. Using the local nature of the gene co-expression we develop algorithms to partition of the metabolic network into local co-regulated modules.
The large-scale structure of genetic networks: design, history, or (mere)
chemistry?
Andreas Wagner
University of New Mexico, Albuquerque, NM
Functional genomics is generating much information about the structure of genetic networks, information that is largely qualitative. How much biology can we learn from such qualitative information? I will address this question in the context of the two well-studied networks of metabolism and protein interactions. Specifically, I will ask whether these networks have their observed structure because this structure provides robustness against mutations. I will also ask whether this structure contains information about the history of these networks and of life itself.
Building Developmental Networks Genome-wide in Drosophila
Kevin White
Yale University, New Haven, CT
Development is controlled by complex molecular networks that are responsible for generating spatial pattern and for controlling the timing of cellular differentiation. I will discuss my laboratory's recent work using gene expression, protein-protein interaction, and protein-DNA interaction data collected on a large scale to analyze Drosophila development. First I will discuss our identification of the targets of homeobox factors that set up the segmental body plan in the early embryo. Second I will describe our work to identify genes involved in the morphological response to the hormone ecdysone, which triggers metamorphosis and coordinates developmental timing. Using these examples, I will discuss some of the strategies and challenges of integrating experimental and computational approaches to delineate the networks that control developmental pattern in space and in time.
| |